语言

 心脏纤维化
心脏纤维化
 z-bio
z-bio
 2025-05-16
2025-05-16
 4875
4875
#心脏纤维化(Cardiac Fibrosis, CF)是心力衰竭、心房颤动等心血管疾病的共同病理基础,表现为心肌成纤维细胞异常增殖迁移及胶原沉积,最终导致心脏功能衰竭。尽管这种疾病在全球范围内高发,但其具体机制尚未完全明确。
今年3月,安徽医科大学第二附属医院陶辉教授团队联合上海交通大学医学院附属新华医院赵健元教授、张野教授团队,在《European Heart Journal》发表了题为“SLC31A1 loss depletes mitochondrial copper and promotes cardiac fibrosis”的研究,首次系统揭示了#线粒体铜耗竭通过糖酵解重编程 及 #m6A 表观遗传通路驱动心脏纤维化的新机制,为 CF 的防治提供了突破性理论依据。

文章由安徽医科大学第二附属医院硕士研究生涂彬、宋凯和上海交通大学医学院博士后周泽宇共同作为第一作者,一起来看看这篇集线粒体铜耗竭+糖酵解+表观遗传(m6A)三大国自然热点于一体的文章是怎么做的吧。
研究背景与科学问题
铜是人体必需的微量元素,参与能量代谢、抗氧化等多种生理过程。此前研究已发现,铜代谢异常与癌症、糖尿病等多种疾病相关,但其在心脏纤维化中的作用仍不清楚。研究人员注意到,在心脏纤维化患者和小鼠模型中,心脏组织的铜含量显著降低,而负责铜转运的基因SLC31A1的表达也明显下降。这提示铜代谢失调可能与心脏纤维化的发生密切相关。
线粒体铜耗竭:心脏纤维化的关键驱动因素
临床与动物模型验证
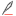
研究团队通过电感耦合等离子体质谱(ICP-MS)发现,在 Ang II/ISO 诱导的小鼠 CF 模型、心肌梗死模型及房颤患者心脏组织中,铜离子含量显著降低,且铜转运蛋白 SLC31A1 表达下调。荧光探针示踪显示,线粒体铜分布异常与线粒体形态碎片化、嵴数量减少直接相关,提示线粒体铜代谢紊乱是 CF 的早期事件。
体外实验表明,Ang II 刺激心脏成纤维细胞可导致线粒体膜电位下降、氧消耗率(OCR)降低、细胞外酸化率(ECAR)升高,糖酵解关键酶 HIF-1α、HK3 表达显著增加。敲低 SLC31A1 进一步加剧线粒体铜耗竭,促进成纤维细胞增殖和胶原沉积;而过表达 SLC31A1 或补充铜离子则逆转这一过程,证实线粒体铜耗竭通过驱动糖酵解重编程促进 CF。
m6A 表观遗传调控网络:SLC31A1 失活的分子机制
DNA 甲基化与转录抑制
机制研究发现,SLC31A1 启动子区存在典型 CpG 岛,其甲基化水平在 CF 模型及房颤患者中显著升高。甲基化 DNA 免疫沉淀(MeDIP)和染色质免疫沉淀(ChIP)实验证实,甲基化 CpG 结合蛋白 MeCP2 通过识别 SLC31A1 启动子甲基化位点抑制其转录,导致铜转运功能缺陷。
m6A 修饰的上游调控作用
通过 m6A 测序和 RNA 结合蛋白分析,团队发现 YTHDF1 作为 m6A 阅读器,特异性识别 MeCP2 mRNA 的 m6A 修饰位点,增强其稳定性并促进翻译。敲低 YTHDF1 可减少 MeCP2 表达,恢复 SLC31A1 转录,从而逆转线粒体铜耗竭和 CF 表型。临床样本验证显示,房颤患者心脏组织中 YTHDF1/MeCP2 表达升高与 SLC31A1 甲基化呈正相关,进一步确认了YTHDF1/MeCP2/m6A 通路在 CF 中的核心作用。
研究团队还在心房颤动(AF)和糖尿病心肌病(DCM)患者的组织样本中验证了上述机制。这些患者的心脏组织中同样存在铜含量降低、SLC31A1表达减少以及MeCP2/YTHDF1水平升高的现象。这表明,针对铜代谢和SLC31A1的调控可能成为治疗心脏纤维化的新策略。
铜补充能否成为疗法?
尽管补充铜看似直观,但研究发现,单纯补铜在SLC31A1缺失的情况下效果有限。因此,未来研究可以聚焦于如何通过基因治疗或表观遗传药物(如DNA去甲基化剂)恢复SLC31A1的功能。此外,探索YTHDF1/MeCP2通路的抑制剂也可能为临床干预开辟新途径。
该研究创新性地将线粒体铜代谢、糖酵解重编程与 m6A 表观遗传三大前沿领域有机整合,揭示了心脏纤维化的新型分子网络。其科学价值不仅在于阐明了 SLC31A1 在心血管疾病中的双重角色,更首次证实 m6A 修饰通过调控金属稳态参与器官纤维化,为心血管表观遗传学研究开辟了新方向。
未来,针对 YTHDF1/MeCP2/SLC31A1 轴的精准干预,有望成为逆转心脏纤维化、改善房颤预后的关键突破点。