Language

 心脏纤维化
心脏纤维化
 z-bio
z-bio
 2025-05-16
2025-05-16
 4820
4820
#Cardiac fibrosis (CF) is a common pathological basis of cardiovascular
diseases such as heart failure and atrial fibrillation, characterized by
abnormal proliferation and migration of myocardial fibroblasts and collagen
deposition, ultimately leading to heart failure. Although this disease is
prevalent globally, its specific mechanism is not yet fully understood.
In March of this year, Professor Tao Hui's team from the Second Affiliated
Hospital of Anhui Medical University, together with Professor Zhao Jianyuan and
Professor Zhang Ye's team from the Xinhua Hospital affiliated with Shanghai Jiao
Tong University School of Medicine, published a study titled "SLC31A1 loss
depletion mitochondrial copper and promotes cardiac fibrosis" in the European
Heart Journal. For the first time, the study systematically revealed a new
mechanism of # mitochondrial copper depletion driving cardiac fibrosis through
glycolysis reprogramming and # m6A epigenetic pathway, providing a breakthrough
theoretical basis for the prevention and treatment of CF.

The article is co authored by Tu Bin and Song Kai, master's students from
the Second Affiliated Hospital of Anhui Medical University, and Zhou Zeyu,
postdoctoral fellow at the School of Medicine of Shanghai Jiao Tong University.
Let's take a look at how this article integrates three major natural hotspots:
mitochondrial copper depletion, glycolysis, and epigenetics (m6A).
Research Background and Scientific Issues
Copper is an essential trace element for the human body, involved in
various physiological processes such as energy metabolism and antioxidant
activity. Previous studies have found that abnormal copper metabolism is
associated with cancer, diabetes and other diseases, but its role in heart
fibrosis is still unclear. Researchers noticed that copper content in heart
tissue was significantly reduced in patients with cardiac fibrosis and mouse
models, and the expression of the gene SLC31A1 responsible for copper transport
was also significantly decreased. This suggests that copper metabolism imbalance
may be closely related to the occurrence of cardiac fibrosis.
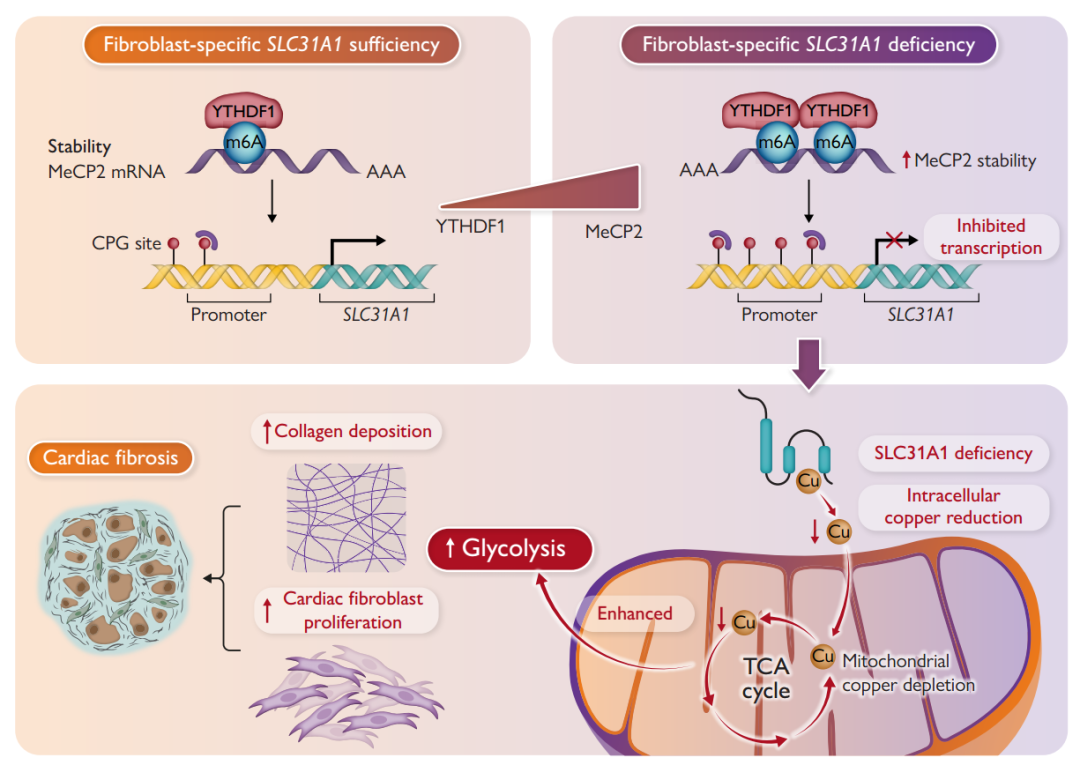
Mitochondrial copper depletion: a key driver of cardiac fibrosis
Clinical and animal model validation
The research team found through inductively coupled plasma mass
spectrometry (ICP-MS) that copper ion content was significantly reduced and
copper transporter SLC31A1 expression was downregulated in Ang II/ISO induced
mouse CF models, myocardial infarction models, and heart tissues of atrial
fibrillation patients. Fluorescence probe tracing showed that abnormal
distribution of mitochondrial copper was directly related to mitochondrial
morphological fragmentation and reduced cristae count, suggesting that
mitochondrial copper metabolism disorder is an early event in CF.
In vitro experiments have shown that Ang II stimulation of cardiac
fibroblasts can lead to a decrease in mitochondrial membrane potential, a
decrease in oxygen depletion rate (OCR), an increase in extracellular
acidification rate (ECAR), and a significant increase in the expression of key
glycolytic enzymes HIF-1 α and HK3. Knocking down SLC31A1 further exacerbates
mitochondrial copper depletion, promotes fibroblast proliferation and collagen
deposition; Overexpression of SLC31A1 or supplementation of copper ions reversed
this process, confirming that mitochondrial copper depletion promotes CF by
driving glycolytic reprogramming.
M6A epigenetic regulatory network: molecular mechanism of SLC31A1
inactivation
DNA methylation and transcriptional repression
Mechanism studies have found that there are typical CpG islands in the
SLC31A1 promoter region, and their methylation levels are significantly elevated
in CF models and atrial fibrillation patients. Methylated DNA
immunoprecipitation (MeDIP) and chromatin immunoprecipitation (ChIP) experiments
have confirmed that the methylated CpG binding protein MeCP2 inhibits
transcription of SLC31A1 by recognizing its methylation site in the promoter,
leading to copper transport dysfunction.
Upstream regulatory effect of m6A modification
Through m6A sequencing and RNA binding protein analysis, the team found
that YTHDF1, as an m6A reader, specifically recognizes the m6A modification site
of MeCP2 mRNA, enhances its stability, and promotes translation. Knocking down
YTHDF1 can reduce MeCP2 expression, restore SLC31A1 transcription, and reverse
mitochondrial copper depletion and CF phenotype. Clinical sample validation
shows that the elevated expression of YTHDF1/MeCP2 in the heart tissue of atrial
fibrillation patients is positively correlated with SLC31A1 methylation, further
confirming the core role of the YTHDF1/MeCP2/m6A pathway in CF.
The research team also verified the above mechanism in tissue samples of
patients with atrial fibrillation (AF) and diabetes cardiomyopathy (DCM). There
is also a decrease in copper content, a decrease in SLC31A1 expression, and an
increase in MeCP2/YTHDF1 levels in the heart tissue of these patients. This
suggests that regulating copper metabolism and SLC31A1 may become a new strategy
for treating cardiac fibrosis.
Can copper supplementation become a therapy?
Although copper supplementation may seem intuitive, research has found that
the effect of copper supplementation alone is limited in the absence of SLC31A1.
Therefore, future research can focus on how to restore the function of SLC31A1
through gene therapy or epigenetic drugs such as DNA demethylating agents. In
addition, exploring inhibitors of the YTHDF1/MeCP2 pathway may also open up new
avenues for clinical intervention.
This study innovatively integrates mitochondrial copper metabolism,
glycolysis reprogramming, and m6A epigenetics, revealing a novel molecular
network of cardiac fibrosis. Its scientific value lies not only in elucidating
the dual role of SLC31A1 in cardiovascular disease, but also in confirming for
the first time that m6A modification participates in organ fibrosis by
regulating metal homeostasis, opening up new directions for cardiovascular
epigenetics research.
In the future, precise intervention targeting the YTHDF1/MecP2/SLC31A1 axis
is expected to become a key breakthrough point in reversing cardiac fibrosis and
improving the prognosis of atrial fibrillation.